Artificial Intelligence, Molecular design, Molecular dynamics, Free energy calculation, Drug design, Drug discovery
Prof. Juyong Lee
- Molecular generative models
- Chemical language model
- Developing computational drug discovery pipeline
- Protein design
Current Research Interests
1. Molecular generative models
We are interested in developing new algorithms to generate novel drug candidates using artificial intelligence and global optimization algorithms. Efficient molecular design algorithms will contribute to accelerate drug discovery process as well as to design novel functional organic materials. The major limitation of existing molecular generative algorithms is that they tend to generate molecules hard to be synthesized. We are trying to develop AI models that consider both optimal properties and synthetic feasibility.
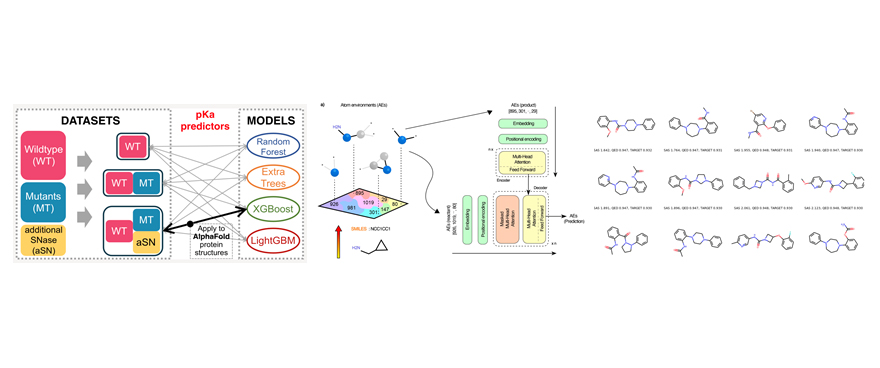
2. Chemical language models
One of the most widely used molecular representation is a text-based representation, such as SMILES, InChI and SELFIES. Chemical language models learn the hidden patterns and laws of molecules in terms of text representation. The models can shed light on the hidden structure of chemical space. Most existing chemical language models rely on the SMILES representation. However, SMILES consists of limited number of tokens. We are pushing the limitations of chemical language models by devising new tokenization scheme and training model architecture.
3. Developing computational drug discovery pipeline
The goal of our research is to develop a comprehensive end-to-end computational drug discovery pipeline using artificial intelligence and physics-based calculations. This pipeline encompasses ultra-fast virtual screening, drug candidate design, binding affinity prediction, retrosynthesis prediction, and ADMET prediction.
4. Protein design
We also utilize many recent protein-structure prediction techniques and generative artificial intelligence to design novel functional proteins. We are also interested in developing antibody-design algorithms.